Pathogenic Variant Analysis in the Esan Population Sample
Group members : Sumia Basunia, Chirag Jain, YooJin Joung, Siulung Ng, Maya Rajan, Ankit Srivastava
Background
The 1000 Genomes Project was the largest international project pursuing an extensive collection of genetic variation. Researchers sequenced the genomes of about 1000 participants, 14 populations taken from all around the world. The database contains a detailed collection of rare variants, including SNPs, structural variants, and indels. The project has ultimately made huge advances in population genetics and comparative genomics today. This data can be used to identify genetic predispositions to cancers and diseases in specific populations compared to the global population. This experiment aims to review identified pathogenic variants found in the Esan population from the 1000 Genomes project.
Objectives
The objective of the project was to analyze different pathogenic variants of genes from 1000 Genomes exome sequencing data. The data used in the analysis was of a male originated in Esan, Nigeria and the observed variants all differed at a single nucleotide, or SNPs. The variants were further studied for their pathogenicity, frequency, evolutionary context and the effect of allelic genotype on affected individuals.
Data description and Pipeline
We selected Esan in Nigeria as the population for this project. From the population, we used the sample with ID HG02938, which came from a male, for our analysis. We downloaded the paired-end sequence read data from the sample data and aligned it to Feb. 2009 assembly of the human genome (hg19, GRCh37) using Burrows-Wheeler Aligner (BWA) [9]. Then, using the result of the alignment, we found the list of variants using SAMtools [10] and BCFtools.

Figure 1 : Pipeline to identify and analyse the pathogenic SNP variants
Annotation & filtering scheme
The VCF file generated using Mpileup had about 370,000 variants. We annotated these variants to study the affected genes, proteins and consequences using Variant Effect Predictor hosted on Ensembl. Using this output, we filtered out the 443 variants which had pathogenic clinical significance. We picked 6 variants out of these 443 variants and cross checked their clinical significance in the dbSNP database.

Table 1: List of six variants studied in this project. Some of these affect multiple transcript variants.
Variants
-
rs6025
Coagulation Factor V (F5) is an essential cofactor of the blood coagulation process. Initially generated in the liver, F5 circulates in the blood plasma until it is converted to an active form by thrombin to initiate coagulation. Defects in the gene result in various diseases related to hemorrhagic diathesis and thrombophilia and also render individuals susceptible to conditions like ischemic stroke, Budd-Chiari syndrome and recurrent abortion.
A missense SNP mutation from G to A at position 1746 (dbSNP id = rs6205) caused an amino acid change from Arginine to Glutamine in the particular variant. Reliability of the genotype was high with sequencing quality of 228; read depth of 74 and BAQ mapping quality of 60. There were no known alternative splice variants of the gene Homo sapiens coagulation factor V (proaccelerin, labile factor). Factor V deficiency as a result of the variant is an autosomal recessive inheritance, occurring at 1 in a million individuals as homozygotes and 1 in 1000 as heterozygotes, or 3.5% of population (Beauchamp et al., 1994). Heterozygous deficiency is generally unrecognized as the gene is haploid sufficient (C/C or C/c), but, although rare, homozygous (c/c) individuals always exhibit symptoms such as clotting time prolongation and bleeding risk (Zehnder et al., 1999). Similar study of mutation has been performed on mouse, observing F5 homologs on chromosome 1 (MGI:2138166).
The mutation occurred in a region evolutionarily conserved among primates, as observed in green in the multiple sequence alignment (Figure 2). The protein structure of coagulation factor V is also shown in Figure 3 and the domain linker where the mutation occurs is represented in teal blue.
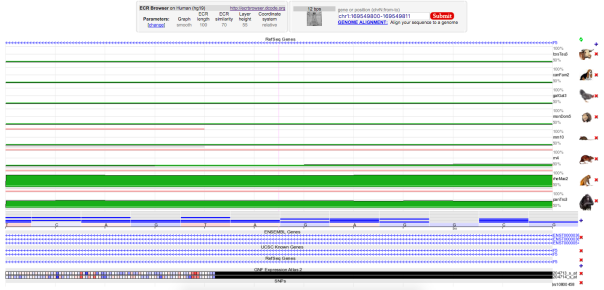
Figure 2 : Enter a captionMultiple Sequence Alignment of various species of animals. The regions indicated by green are indicative of evolutionary conserved region for corresponding animal species. Selected region for mutation(rs6025) was conserved in rheMac2 and panTro3.

Figure 3: The protein structure of Coagulation Factor 5. The region of domain linker where the mutation occurs is represented in teal blue.
-
rs1061170
Complement factor H (CFH) gene is a member of the regulator of complement activation gene that positioned at Homo sapiens chromosome 1. It encodes a serum glycoprotein, CFH, that circulates in human plasma and involves in the regulation of the alternative complement pathway, which is part of the immunity system, in order to prevent damaging the host cells and guiding the complement system to target any harmful elements, including pathogens.
dbSNP shows that a missense SNP mutation from C to T at mRNA position 1444 (dbSNP id = rs1061170) causes an amino acid (AA) change from histidine to tyrosine at AA sequence position 402. However, other sources, such as ClinVar and the studies on OMIM suggested that the mutation at the same position is actually a T to C mutation, resulting a single AA change from tyrosine to histidine. Thus, the SNP mutation provided by dbSNP is incorrect. In addition, the genotype is proven to be reliable based on the sequencing quality (= 228), mapping quality (= 60), and read depth (= 48). There are two alternative splice variants found, named CFH transcript variant 1 (GI: 184172390) and 2 (GI: 184172391), which are translated into CFH isoforms A (GI: 62739186) and B (GI: 62739188), respectively. The 3D protein structure is shown in Figure 5 with the highlighted mutation position (Yellow). The mutation site is proven to be 100% conserved in panTro3 and 50% conserved in rheMac2 and canFam2 (Figure 4).

Figure 4 : Multiple Sequence Alignment of various species of animal showing the selected mutation (rs1061170) is evolutionary conserved in panTro3 and partially conserved in rheMac2 and canFam2.
The T to C SNP mutation can lead to basal laminar drusen and age-related macular degeneration. Although no animal model was mentioned in the studies of basal laminar drusen and age-related macular degeneration (ARMD) diseases, other genetic diseases, such as CHF deficiency, that caused by different mutations in this allele were studied using mice (Pickering et al., 2002; Pickering et al., 2006; Coffey et al., 2007) and Norwegian Yorkshire pig model (Hogasen et al., 1995). For the basal laminar drusen, it is an autosomal recessive disease, meaning that two copies of an abnormal gene must be present in order for the disease to develop (Boon et al.,2008). Therefore, people with heterozygous alleles or normal homozygous alleles should not have basal laminar drusen. For ARMD, a meta-analysis suggested that the mutated allele in either heterozygous or homozygous state can develop ARMD (Conley et al., 2006). The details of prevalence for both diseases are not clear.

Figure 5 : Enter a caption3D protein structure of chain A, solution structure of human CFH in 50 Mm NaCl buffer. The location of variant amino acid is shown in yellow.
-
rs1800566
NAD(P)H dehydrogenase (NQO1) gene that codes for a protein used to catalyzes various quinones such as vitamin K menadione and phyloquinone (Jaiswal et al. 1988). This specific gene from the quinone family codes from cytoplasmic 2-electron reductase ultimately preventing the production of radical species in the body. Without sufficient NQO1 proteins, the body may suffer from benzene toxicity. NQO1 is found on chromosome 16 and contains 6 coding exons over about 20 kb (Jaiswal et al. 1988).
A missense mutation from C to T at 4 sites (dbSNP id = rs1800566), as the NAD(P)H dehydrogenase is made up of 4 NQO1 proteins. The mRNA positions are 750, 648, 636, and 572 for quinone 1 transcript variant 1, 2, 3, and 4 respectively. The dbSNP database shows an allele change from CCT to TCT at all 4 positions mentioned, ultimately causing an amino acid change from Proline to Serine at these sites (Figure 7). The reliability of this genotype was also high with a sequencing quality of 222 and a read depth of 103 (MQ = 60). As seen in Figure 6, the multiple sequence alignment shows the conservation of this variant across multiple species. There are four alternative splice variants found, named NQO1 transcript variant 1 to 4 (GI: 70995356, 70995395, 70995421, 554790419 respectively). This mutation can lead to benzene toxicity and poor survival rates after chemotherapy in leukaemia and breast cancer.
Although no animal model was mentioned in reviewing the survival rates post-chemotherapy in leukemia, studies have shown that mice lacking the NQO1 gene exhibited increased toxicity when administered menadione, a common vitamin K supplement (Radjendirane et al. 1998) .Previous studies have seen the primary effects of NQO1 variants in benzene toxicity (Moran et al. 1999, Rothman et al. 1997). This variant has been shown to have the highest prevalence at 20% in Asian populations and therefore, more studies were pursued in Asian subpopulations (Kelsey et al. 1997). In a more recent study, the common missense variant was found to be a strong predictor of poor survival in women with breast cancer, especially homozygote patients (Fagerholm et al. 2008). As these research studies have been thoroughly followed up, there is no sign that this variant is non-pathogenic.
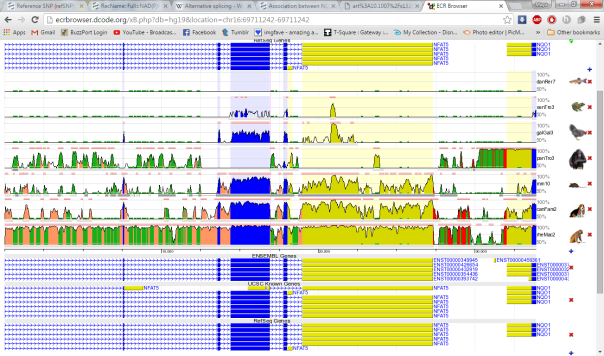
Figure 6 : Multiple Sequence Alignment (MSA) of various species of animal showing the selected mutation (rs1800566) is shown to be heavily conserved in macaque, dog, mice and chimpanzee. Limited conservation is seen against zebrafish, frog and chicken.
Multiple Sequence Alignment (MSA) of various species of animal showing the selected mutation (rs1800566) is shown to be heavily conserved in macaques, dogs, mice, and chimpanzees. Limited conservation is seen in zebrafish, frogs, and chickens.
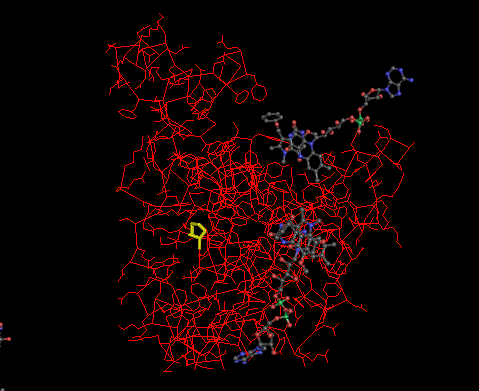
Figure 7 : Enter a caption3D Protein structure of Chain A, Complex of Human Recombinant Nad(P)h:quinone Oxidoreductase Type 1 with 5-Methoxy-1,2-Dimethyl-3-(Phenoxymethyl)indole-4,7-Dione. The location of the variant mutation can be seen in yellow. 3D model is generated via Cn3D.
-
rs846664
Taste receptor, type 2, member 16 (TAS2R16) gene is located in Human (Homo sapiens) chromosome 7. It encodes a bitter taste receptor protein, which is specifically expressed by taste receptor cells of the tongue and palate epithelia and functions as a bitter taste receptor. A missense mutation from Thymine to Guanine (AAT ⇒ AAG), at position 582, causes an amino acid change from Asparagine to Lysine (N => K), at position 172, in this variant (dbSNP ID = rs846664). Figure 9 shows the position of the variant in the 3D protein structure. The genotype is reliable with read depth of 113, phred-scaled sequencing quality score of 222, and mapping quality of 60. The gene lacks introns therefore there are no splice variants affected by this variant. Multiple sequence alignment of the region (shown in Figure 8) suggests that the change was at a site which was evolutionarily conserved in Rhesus (Macaca mulatta) and Chimp (Pan troglodytes).
Multiple studies on this variant have been performed on Human models which have linked it to susceptibility to alcohol dependence (Zehnder et al. 1999, Eerdewegh et al. 1998, Tatiana et al. 2000, Anthony et al. 2006). The variant is shown to be common in African Americans, with 45% of the population carrying the allele, whereas it is uncommon in European Americans (Tatiana et al. 2000, Anthony et al. 2006). Other studies have suggested the possibility of a Central African of this allele (Wang et al. 2007, Nicole et al. 2005). Another study on Human models has linked it to Beta-glycopyranoside tasting (Nicole et al. 2005).
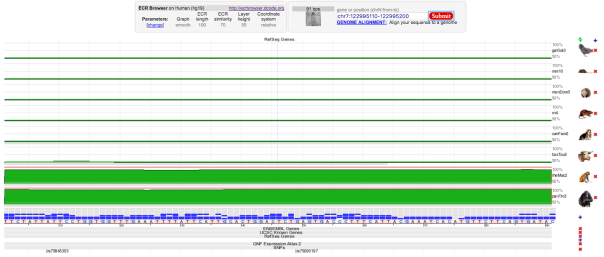
Figure 8 : Multiple Sequence Alignment of various species of animal showing the selected mutation (rs846664) was evolutionary conserved in rheMac2 and panTro3

Figure 9: Part of the protein structure of Taste receptor type 2 member 16. The location of the variant amino acid is shown in purple, inside the region coded with yellow.
-
rs1799983
rs1799983 variant is associated with the NOS3 gene responsible for the synthesis of nitric oxide free radical. This variant is alteration of the protein sequence at position 298 from D (Asparate) into E (Glutamate). The genomic loci of this SNP is 150,696,111 in chromosome 7 of the GRCh37 assembly. The sequencing depth at this position is 25 and the BAQ mapping quality is 60 which implies that this SNP call can be assumed to be reliable. There are 5 known transcript variants affected by this variant (NM_000603.4, NM_001160109.1, NM_001160110.1, NM_001160111.1, XP_006716065). The evolutionary conservation has been shown in Figure 10 using the ECR browser, where we see that the region is conserved among the closely related species: rhesus monkey, chimpanzee but not with the distant taxonomic species such as chicken.
Using clinvar, we explored the recent clinical studies which report the effect of this variant. There is evidence of this SNP responsible for building the susceptibility towards coronary artery spasm, hypertension and ischemic heart disease. OMIM database contains a list of the related studies and experiments. It has been shown that Nitric oxide (NO) accounts for the biological activity of endothelium-derived relaxing factor (EDRF). EDRF is crucial for our body to maintain the blood flow as it inhibits muscle contraction and platelet aggregation. (Yoshimura et al. 1998) showed that the occurrence of this variant was more frequent in patients with coronary spasm. (Tanus-Santos et al. 2001) studied the demographic distribution of this variant where they reported that this variant was more common in Caucasians (34.5%) compared to African Americans (15.5%) or Asians (8.6%). 3D structure of the protein with the variant circled is shown in Figure 11.
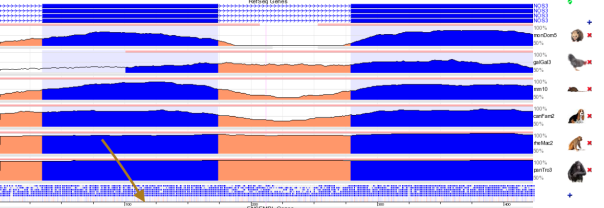
Figure 10 : Multiple Sequence Alignment of multiple animals showing that the selected mutation (rs1799983) is evolutionary conserved in rhesus monkey and chimpanzee. Brown arrow points at the location of the variant within the alignment.
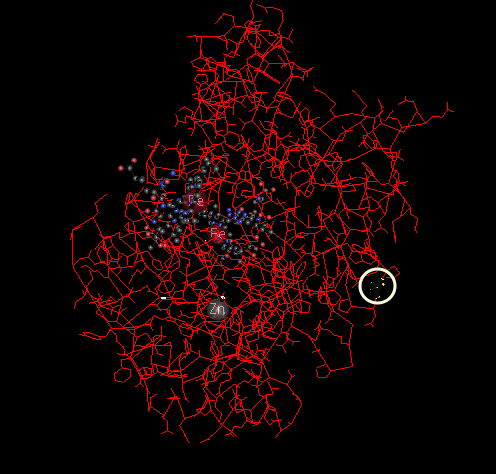
Figure 11 : Chain A, Human Endothelial Nitric Oxide Synthase with Arginine Substrate (3NOS_A). Location of the variant amino acid is highlighted using a while circle.
-
rs3970559
Proline dehydrogenase is part of a group of enzymes referred to as oxidoreductases. Proline dehydrogenase functions to catalyze a reaction between L-proline and ubiquinone to yield two products of S-1-pyrroline-5-carboxylate and ubiquinol. Found in the brain, kidneys and liver, proline dehydrogenase function inside the mitochondria of the cell to facilitate energy transfer.
In a variant of proline dehydrogenase, a missense single nucleotide polymorphism (SNP) mutation from R to C (dbSNP id = rs3970559) produces an amino acid change from Arginine to Cysteine. The single nucleotide polymorphism occurs when a cytosine changes to a thymine. Characteristics of this SNP include a sequencing quality of 222, a mapping quality of 5 and a depth of 118. The mRNA positions that cause a residue change from Arginine to Cysteine include 952, 1292, 1561, and 1146 leading to alternative spliced transcript variants in X3,X2,1 and 2, respectively.
The pathogenic variant disrupts enzyme ability to perform proline metabolism. Phenotypic consequences of this pathogenic variant include proline dehydrogenase deficiency and schizophrenia.Multiple papers found a connection between this pathogenic variant and hyperprolinemia and schizoaffective disorder (Jacquet 2002, Jacquet 2005). The frequency of this pathogenic variant is GO-ESP 0.00915 (A) and GMAF 0.02280 (A). Population geneticists have yet to study the prevalence of this pathogenic variant in the human population and extrapolate risk factors for certain ethnic or racial groups. According to OMIM, ClinVar, and dbSNP database, there is no evidence of any exceptions where this variant has been reported to be non-pathogenic.
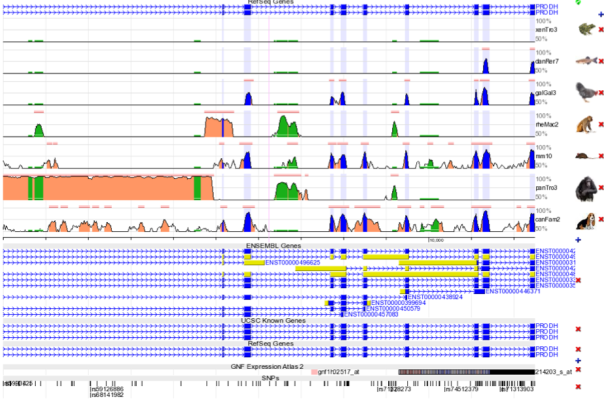
Figure 12 : Multiple Sequence Alignment for the evolutionary conserved site where the missense single nucleotide polymorphism (SNP) mutation from R to C (dbSNP id = s3970559) occurs that produces an amino acid change from Arginine to Cysteine. Within the species list, this site is primarily conserved in the rheMac2 and panTro3 as seen above.
Figure 13: A 3D rendering of the proline dehydrogenase. No rendering for this particular pathogenic variant is available.
Discussion and Conclusion
In this project, we used multiple softwares such as samtools, variant effect predictors to perform the reliable SNP calling and annotation. This was followed by studying the significance and literature review about the variants using multiple databases and data archives like dbSNP, OMIM and clinVar.
6 filtered variants (dbSNP id: rs6025, rs1061170, rs1800566, rs846664, rs1799983, and rs3970559) out of 443 were selected for the SNP analysis. These SNP mutations have been proven to be a missense mutations that can cause human genetic disorders by changing a single amino acid. These pathogenic variants lead to various diseases described above.
Modeling these pathogenic variants in different population will allow us to predict the risk factors associated with each race or ethnic group. In addition, these diseases can be predicted in the future generation if newborns carry the pathogenic variants.
References
- Jaiswal, A. K., McBride, O. W., Adesnik, M., Nebert, D. W. Human dioxin-inducible cytosolic NAD(P)H:Menadione oxidoreductase: cDNA sequence and localization of gene to chromosome 16. J. Biol. Chem. 263: 13572-13578, 1988.
- Radjendirane, V., Joseph, P., Lee, Y.-H., Kimura, S., Klein-Szanto, A. J. P., Gonzalez, F. J., Jaiswal, A. K.Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J. Biol. Chem. 273: 7382-7389, 1998.
- Moran, J. L., Siegel, D., Ross, D. A potential mechanism underlying the increased susceptibility of individuals with a polymorphism in NAD(P)H:quinone oxidoreductase 1 (NQO1) to benzene toxicity. Proc. Nat. Acad. Sci. 96: 8150-8155, 1999.
- Rothman, N., Smith, M. T., Hayes, R. B., Traver, R. D., Hoener, B., Campleman, S., Li, G. L., Dosemeci, M., Linet, M., Zhang, L., Xi, L., Wacholder, S., Lu, W., Meyer, K. B., Titenko-Holland, N., Stewart, J. T., Yin, S., Ross, D. Benzene poisoning, a risk factor for hematological malignancy, is associated with the NQO1 609C-T mutation and rapid fractional excretion of chlorzoxazone. Cancer Res. 57: 2839-2842, 1997.
- Kelsey, K. T., Ross, D., Traver, R. D., Christiani, D. C., Zuo, Z. F., Spitz, M. R., Wang, M., Xu, X., Lee, B. K., Schwartz, B. S., Wiencke, J. K. Ethnic variation in the prevalence of a common NAD(P)H quinone oxidoreductase polymorphism and its implications for anti-cancer chemotherapy. Brit. J. Cancer 76: 852-854, 1997.
- Fagerholm, R., Hofstetter, B., Tommiska, J., Aaltonen, K., Vrtel, R., Syrjakoski, K., Kallioniemi, A., Kilpivaara, O., Mannermaa, A., Kosma, V.-M., Uusitupa, M., Eskelinen, M., Kataja, V., Aittomaki, K., von Smitten, K., Heikkila, P., Lukas, J., Holli, K., Bartkova, J., Blomqvist, C., Bartek, J., Nevanlinna, H.NAD(P)H:quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nature Genet. 40: 844-853, 2008.
- Beauchamp, N. J., Daly, M. E., Hampton, K. K., Cooper, P. C., Preston, E., Peake, I. R. “High prevalence of a mutation in the factor V gene within the U.K. population: relationship to activated protein C resistance and familial thrombosis.” Brit. J. Haemat. 88: 219-222, 1994.
- Zehnder, J. L., Hiraki, D. D., Jones, C. D., Gross, N., Grumet, F. C. “Familial coagulation factor V deficiency caused by a novel 4 base pair insertion in the factor V gene: factor V Stanford.” Thromb. Haemost. 82: 1097-1099, 1999. Note: Erratum: Thromb. Haemost. 82: XII only, 1999.
- Van Eerdewegh, Paul, Tatiana Foroud, Victor Hesselbrock, Marc A. Schuckit, Kathleen Bucholz, Bernice Porjesz, Ting-Kai Li et al. “Genome-wide search for genes affecting the risk for alcohol dependence.” American Journal of Medical Genetics (Neuropsychiatric Genetics) 81 (1998): 207-215.
- Foroud, Tatiana, Howard J. Edenberg, Alison Goate, John Rice, Leah Flury, Daniel L. Koller, Laura J. Bierut et al. “Alcoholism susceptibility loci: confirmation studies in a replicate sample and further mapping.” Alcoholism: Clinical and Experimental Research 24, no. 7 (2000): 933-945.
- Hinrichs, Anthony L., Jen C. Wang, Bernd Bufe, Jennifer M. Kwon, John Budde, Rebecca Allen, Sarah Bertelsen et al. “Functional variant in a bitter-taste receptor (hTAS2R16) influences risk of alcohol dependence.” The American Journal of Human Genetics 78, no. 1 (2006): 103-111.
- Wang, Jen C., Anthony L. Hinrichs, Sarah Bertelsen, Heather Stock, John P. Budde, Danielle M. Dick, Kathleen K. Bucholz et al. “Functional Variants in TAS2R38 and TAS2R16 Influence Alcohol Consumption in High‐Risk Families of African‐American Origin.” Alcoholism: Clinical and Experimental Research 31, no. 2 (2007): 209-215.
- Soranzo, Nicole, Bernd Bufe, Pardis C. Sabeti, James F. Wilson, Michael E. Weale, Richard Marguerie, Wolfgang Meyerhof, and David B. Goldstein. “Positive selection on a high-sensitivity allele of the human bitter-taste receptor TAS2R16.” Current Biology 15, no. 14 (2005): 1257-1265.
- Bufe, Bernd, Thomas Hofmann, Dietmar Krautwurst, Jan-Dirk Raguse, and Wolfgang Meyerhof. “The human TAS2R16 receptor mediates bitter taste in response to β-glucopyranosides.” Nature genetics 32, no. 3 (2002): 397-401.
- Pickering, M. C., Cook, H. T., Warren, J., Bygrave, A. E., Moss, J., Walport, M. J., Botto, M. “Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H.” Nature Genet. 31: 424-428, 2002.
- Pickering, M. C., Warren, J., Rose, K. L., Carlucci, F., Wang, Y., Walport, M. J., Cook, H. T., Botto, M. “Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice.” Proc. Nat. Acad. Sci. 103: 9649-9654, 2006.
- Coffey, P. J., Gias, C., McDermott, C. J., Lundh, P., Pickering, M. C., Sethi, C., Bird, A., Fitzke, F. W., Maass, A., Chen, L. L., Holder, G. E., Luthert, P. J., Salt, T. E., Moss, S. E., Greenwood, J. “Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction.” Proc. Nat. Acad. Sci. 104: 16651-16656, 2007.
- Boon, C. J. F., Klevering, B. J., Hoyng, C. B., Zonneveld-Vrieling, M. N., Nabuurs, S. B., Blokland, E., Cremers, F. P. M., den Hollander, A. I. “Basal laminar drusen caused by compound heterozygous variants in the CFH gene.” Am. J. Hum. Genet. 82: 516-523, 2008.
- Conley, Yvette P., Johanna Jakobsdottir, Tammy Mah, Daniel E. Weeks, Ronald Klein, Lewis Kuller, Robert E. Ferrell, and Michael B. Gorin. “CFH, ELOVL4, PLEKHA1 and LOC387715 genes and susceptibility to age-related maculopathy: AREDS and CHS cohorts and meta-analyses.” Human Molecular Genetics 15 (21):3206-3218, 2006.
- Hogasen, K., Jansen, J. H., Mollnes, T. E., Hovdenes, J., Harboe, M. “Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiency.” J. Clin. Invest. 95: 1054-1061, 1995.
- Yoshimura, M., Yasue, H., Nakayama, M., Shimasaki, Y., Sumida, H., Sugiyama, S., Kugiyama, K., Ogawa, H., Ogawa, Y., Saito, Y., Miyamoto, Y., Nakao, K. A missense glu298asp variant in the endothelial nitric oxide synthase gene is associated with coronary spasm in the Japanese. Hum. Genet. 103: 65-69, 1998.
- Tanus-Santos, J. E., Desai, M., Flockhart, D. A. Effects of ethnicity on the distribution of clinically relevant endothelial nitric oxide variants. Pharmacogenetics 11: 719-725, 2001.
- Jacquet, H. PRODH Mutations and Hyperprolinemia in a Subset of Schizophrenic Patients. Human Molecular Genetics, 2002, 2243-249.
- Jacquet, H. Hyperprolinemia is a risk factor for schizoaffective disorder. Molecular Psychiatry, 2005, 479-485
Posted on December 7, 2015, in Exome Analysis, Uncategorized. Bookmark the permalink. Leave a comment.

Leave a comment
Comments 0