Author Archives: etnorris
Exome Analysis of Individual from Han Chinese Population
BIOL 6150: Emily Norris, Eshaw Vidyaprakash, Krutika Satish Gaonkar, Lori Gladney
Objective:
The aim of this project was to find variants in the HG00569 exome data set from the 1000 Genomes Project. The individual chosen was from the Han Chinese population. Their genome was sequence using the Illumina Platform.
Methods:
Variant Calling and Filtering
We used SAMTools mpileup to generate the BCF file from the .bam file downloaded from the 1000 Genomes Project.
Command: samtools mpileup -uf fastafile.fa bamfile.bam | bcftools view -bvcg > output.bcf
We filtered (with a threshold depth of 100) the .bcf file then converted it to a .vcf file.
Command: bcftools view output.bcf | vcfutils.pl varFilter -D100 > output.vcf
ANNOVAR
We used ANNOVAR to annotate our genes with the following commands:
Gene Annotation: annotate_variation.pl -geananno out.vcf.annovar hmandb/ Filter Based Annotation: perl annotate_variation.pl -filter -dbtype 1000g2012apr_all -buildver hg19 out.vcf.annovar humandb/ -maf 0.05 perl annotate_variation.pl -filter -dbtyp 1000g2012apr_all -buildver hg19 out.vcf.annovar humandb/
Gene-Talk
The .vcf file generated from the above command was given as input and then analyzed for disease-related variants. We set the following filter parameters to reduce the number of variants
Functional Filter: Nonsynonymous, missense, nonsense; Synonymous Population Frequency: < 0.1%
8 variants were chosen manually based on their disease causing probability.
SNP nexus
The information for each variant was loaded into SNP nexus to obtain its Sift and PolyPhen values along with any associations to the Han Chinese population and/or diseases. The Genetic Association Database was used to obtain information on any diseases related to the SNPs.
(ps)2
(ps)2 was used to generate structures for the protein product associated with each variant.
Variants:
HLA-B (Krutika Satish Gaonkar)
HLA class I histocompatibility antigen, B-27 alpha chain
Variant: rs1071816
Protein: P03989 (1B27_HUMAN)
We observed a single nucleotide polymorhism in the HLA-B gene (T—A).
This polymoprhism was predicted in SNP-nexus to be benign (PolyPhen2) and tolerable (SIFT).

The SNP resulted in an amino acid change a position 91 from Tyrosine (Y) to Serine (S) and causesloss of immune function leading to infections of the skin and cancer like leukemia.

The mutation of the tyrosine to serine might cause the change in the helix which potentially causes the polymorphism in the protein. The protein works as a marker for Stevens–Johnson syndrome as concluded from the studies conducted on an individual in the Han population with this disease.( Chung et al) This disease is induced by carbamazepine, a drug commonly prescribed for the treatment of seizures. Carbamazepine causes various forms of hypersensitivity reactions, ranging from maculopapular exanthema to severe blistering reactions.

The genes in the interaction are all involved in major histoncompatibility complex as transactivators and accessory proteins. This complex is essential in presenting the antigen as a foreign particle to the body’s immune system. Inactivation of these complexes due to the mutation may cause decrease in efficiency of the immune system thus making the body more prone to infections and cancer. The HLA-B*1502 allele has been shown to be strongly correlated with carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS-TEN) in the Han Chinese and other Asian populations but not in European populations.(McCormack et. all)
References:
- http://www.ncbi.nlm.nih.gov/pubmed/15057820 ; Chung WH, Hung SI, Hong HS, Hsih MS, Yang LC, Ho HC, Wu JY, Chen YT. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004 Apr 1;428(6982): 486.
- http://europepmc.org/abstract/MED/21428769/reload=0;jessionid=TM6hOP9FsbrEkZ61|U3p.38 ; McCormack M, Alfirevic A, Bourgeois S, Farrell JJ, Kasperavičiūtė D, Carrington M, Sills GJ, Marson T, Jia X, de Bakker PI, Chinthapalli K, Molokhia M, Johnson MR, O’Connor GD, Chaila E, Alhusaini S, Shianna KV, Radtke RA, Heinzen EL, Walley N, Pandolfo M, Pichler W, Park BK, Depondt C, Sisodiya SM, Goldstein DB, Deloukas P, Delanty N, Cavalleri GL, Pirmohamed M. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. The New England Journal of Medicine [2011, 364(12):1134-1143]
PAWR (Kruitka Satish Gaonkar)
PRKC apoptosis WT1 Regulator
Variant: rs2307220
Protein: Q96IZ0 (PAWR_HUMAN)
We observed a single nucleotide variation in the PAWR gene (A—T,). This was predicted by SNP-nexus to be disease causing. The SNP resulted in a change in amino acid from Isoleucine to Glutamate. The structure was not available so we used PS2P v2 to obtain the structure by choosing a template with the lowest e value. Loss in gene function leads to apoptosis, transcription failure, and transcription regulation.


Wang et. all, screened for mutations at the 5′ untranslated region (5’UTR) and all the exonic regions of the PAWR gene in a sample of Han Chinese schizophrenic patients from Taiwan. They identified two missense single nucleotide polymorphisms (SNPs) that are in P78R at exon 2 and I199M at exon 3, respectively.

Wilms tumor 1 (WT1); transcription factor plays an important role in cellular development and cell survival. WT1, regulator gene (PAWR) encodes the human homolog of Par-4 protein. Prostate apoptosis response 4 (Par-4) is a leucine zipper containing protein originally found to be involved with the regulation of cancer cell survival through apoptosis (Ranganathan and Rangnekar, 2005). Therefore a change in the protein might lead to inactivation of this protein-protein interaction leading to apoptosis of the cell. And from previous studies we have found that apoptosis plays a role in the pathophysiology of schizophrenia, and in recent years studies have emerged to addressed this issue. (Catts et al., 2006)
References:
- http://www.ncbi.nlm.nih.gov/pubmed/18281137 ; Ling-Hsuan Wanga, Jen-Yeu Chenb, Ying-Jay Lioub, Ying-Chieh Wangb, I-Ching Laib, Ding-Lieh Liaoc, Chia-Hsiang Chena (2008)Association of missense variants of the PRKC, apoptosis, WT1, regulator (PAWR) gene with schizophrenia Progress in Neuro-Psychopharmacology and Biological Psychiatry. 32,3:p 870-875
- P. Ranganathan, V.M. Rangnekar Regulation of cancer cell survival by Par-4 Ann N Y Acad Sci, 1059 (2005), pp. 76–85
-
V.S. Catts, S.V. Catts, J.J. McGrath, F. Féron, D. McLean, E.J. Coulson et al.Apoptosis and schizophrenia: a pilot study based on dermal fibroblast cell lines Schizophr Res, 84 (2006), pp. 20–28
HLA-DQA1 (Lori Gladney)
Major Histocompatibility Complex, Class II, DR Beta 1
Protein: P04229 (2B11_HUMAN)
HLA-DQA1, alpha chain , belongs to the HLA class II molecule consisting of an alpha (DQA) and a beta chain (DQB).This molecule presents to the immune system peptides that are derived from extracellular proteins and is expressed in antigen presenting cells ( B lymphocytes, dendritic cells, and macrophages).
The mutation in DQA1 is a single nucleotide polymorphism (C–T), resulting in amino acid change at position 70 of Arginine (R) to Tryptophan(W) , causing a missense mutation or an incorrect amino acid substitution. In fact, this make sense since Tryptophan is a larger amino acid than arginine, it could lead to steric hindrance. This is likely a protein that is non- functional or has decreased function. SNP-nexus predicts this mutation as to be damaging. From the SNP-nexus association studies, many different disorders are associated to this mutation, notably autoimmune disorders (lupus), cardiomyopathy, allergic rhinitis, cancer, psoriasis, asthma, endometriosis, and diabetes; all in the Chinese population alone.

HLA-DRB5 (Lori Gladney)
Major Histocompatibilty Complex, Class II, DR Beta 5
Protein: Q30154 (DRB5_HUMAN)
HLA-DRB5 belongs to the HLA class II molecules consisting of an alpha (DRA) and beta (DRB) chain. This molecule presents to the immune system peptides that are derived from extracellular proteins and is expressed in antigen presenting cells (B lymphocytes, dendritic cells, and macrophages).
The mutation in DRB5 is a single nucleotide polymorphism (G–A), resulting in amino acid change at position 33 of Arginine (R) to an un-identified amino acid (X) , causing a stop gain (non-sense mutation) in the protein sequence. This is likely a premature stop and thus a protein that is nonfunctional or has severely impaired function. SNP-nexus associates this mutation with narcolepsy and echinococcosis infection in the Chinese population. Only one crystal structure is available which includes the chain DRB5.
References:
- http://www.ncbi.nlm.nih.gov/pubmed/23813219
- http://www.jimmunol.org/content/181/8/5473.long
- http://www.ncbi.nlm.nih.gov/pubmed/11798718?
- http://www.ncbi.nlm.nih.gov/pubmed/16225776?
- http://www.ncbi.nlm.nih.gov/pubmed/16225776?
- http://www.ncbi.nlm.nih.gov/pubmed/15009387?
- http://www.nature.com/ng/journal/v45/n2/full/ng.2518.html?WT.ec_id=NG-201302
LEPRE1 (Emily Norris)
Leucine Proline-Enriched Proteoglycan (Leprecan) 1
Variant: rs3738501
Protein: Q32P28 (Prolyl 3-hydroxylase 1)
LEPRE1 is a protein-coding gene located on chromosome 1. It encodes an enzyme that is a member of a family of collagen prolyl hydroxylases required for collagen biosynthesis, folding, and assembly1. It is known that two variants of this gene exist: GROS1S and GROS1L2. GROS1L is considered to be the canonical version of the gene with an N-terminal signal sequence, RGD motif, leucine zipper, C-terminal endoplasmic reticulum retrieval sequence, N-glycosylation site, and several glycosaminoglycan acceptor sites. GROS1S is C-terminally truncated and lacks the leucine zipper present in GROS1L. Mutations in this gene are associated with osteogenesis imperfect type VII. This disease is characterized by abnormal bone fragility and low bone mass – both of which can lead to susceptibility to fractures.
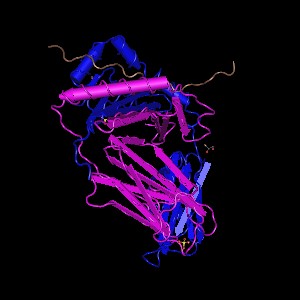
The mutation that occurs in the variant present in our data is a guanine to cytosine missense mutation. It causes a nonsynonymous amino acid change of proline (P) to arginine (R) which alters the structure of the protein.



Normal Q32P28 structure (left) and Altered variant rs3738501 Q32P28 structure (right)
This variant has a minor allele frequency of C = 0.002/4. Both Sift and PolyPhen predict, with high confidence, the P -> R mutation to be damaging. Mutation Taster3 also predicts the variant to be disease causing due to the amino acid change.

Zhang, et al. have shown an association between LEPRE1 mutants and osteogenesis imperfecta in the Chinese population4. They failed to find any LEPRE1 mutations in healthy subjects. Patients with homozygous mutations of LEPRE1 often exhibit lethal types of osteogenesis imperfecta. The HapMap Project confirms the findings of the disease association in the Chinese populations.
References:
- Vranka, J.A., Sakai, L.Y., Bachinger, H.P. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 279: 23615-23621, 2004.
- http://omim.org/entry/610339
- http://www.mutationtaster.org
- Zhang ZL, Zhang H, Ke YH, Yue H, Xiao WJ, Yu JB, Gu JM, Hu WW, Wang C, He JW, Fu WZ. The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J Bone Miner Metab. 301(1): 69-77, 2012.
CACNA1B (Emily Norris)
Calcium Channel, Voltage-Dependent, N Type, Alpha 1B Subunit
Variant: rs4422842
Protein: Q00975 (CAC1B_HUMAN)
CACNA1B encodes the protein for the pore-forming subunit of an N-type voltage-dependent calcium channel. This channel type controls neurotransmitter release from neurons. There have been two variants found for this gene1. According to the Genetic Association Database, these variants are associated with schizophrenia and bipolar disorder.
The mutation that occurs in our variant is a guanine to cytosine shift that causes a nonsynonymous mutation of asparagine (N) to lysine (K) in the protein.

The mutation occurs at amino acid position 167. With high confidence, Sift predicts this variant to be damaging. Mutation Taster also predicts that this is disease causing. Polyphen predicts it to be either “probably damaging” or benign.

Through genome wide association studies, the presence of mutated CACNA1B genes has been associated with schizophrenia and bipolar disorder2. Alterations in copy numbers variants have also been associated with schizophrenia3. The pathways involved in these diseases are the same as those in that CACNA1B is present in.

When alterations are made to the CACNA1B gene, and thus the resulting protein, it is no longer able to make the correct associations with other proteins, leading to the diseased states.

References:
- genecards.org/cgi-bin/carddisp.pl?gene=CACNA1B
- Moskvina V, Craddock N, Holmans P, Nikolov I, Pahwa JS, Green E; Wellcome Trust Case Control Consortium, Owen MJ, O’Donovan MC. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry. 14(3):252-260, 2009.
- Glessner JT, Reilly MP, Kim CE, Takahashi N, Albano A, Hou C, et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc Natl Acad Sci USA. 107(23):10584-10589, 2010.
ETFDH (Eshaw Vidyaprakash)
Electron Transfer Flavoprotein Dehydrogenase
Variant: rs121964954
Protein: Q16134 (ETFD_HUMAN)
ETFDH exists in the mitochondrial matrix as a heterodimer of alpha and beta subunit. It contains 1 flavin adenine dinucleotide and 1 adenosine 5-prime monophosphate per heterodimer1.
The variant of ETFDH found in our dataset is disease causing, containing a single nucleotide variation of G to A. This causes an amino acid change from Alanine (A) to Threonine (T) which leads to the diseased state of the gene.


Both Sift and Polyphen predict that this variant is damaging and disease related. Mutations in the protein lead to altered interactions with other proteins, influencing the biochemical pathways it is involved in.


The mutation is common in Han Taiwanese who suffer from late onset Lipid Storage Myopathies (LSMs)2. The mutation is thought to have originated in South China even though an earlier analysis did not reveal the specific haplotype associated with the mutation.[reference] It has been shown that the presence of ETFDH mutations can help to diagnose Multiple Acyl-CoA Dehydrogenase Deficiency (MADD). Due to large variability in phenotype in contrast to the genetic homogeneity, it is thought that ETFDH, alone, does not cause the disease, but rather environment plays a role as well.
References:
- Gempel, K., Topaloglu, H., Talim, B., Schneiderat, P., Schoser, B. G. H., Hans, V. H., Palmafy, B., Kale, G., Tokatli, A., Quinzii, C., Hirano, M., Naini, A., DiMauro, S., Prokisch, H., Lochmuller, H., Horvath, R. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 130: 2037-2044, 2007.
- Lan, M.-Y., Fu, M.-H., Liu, Y.-F., Huang, C.-C., Chang, Y.-Y., Liu, J.-S., Peng, C.-H., Chen, S.-S. High frequency of ETFDH c.250G-A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin. Genet. 78: 565-569, 2010.
ZBTB40 (Eshaw Vidyaprakash)
Zinc finger and BTB domain containing protein 40
Variant: rs7524102
Protein: Q1RMZ5 (Q1RMZ5_HUMAN)
ZBTB40 is involved in transcription regulation and bone mineralization. It involves alternative splicing with repeats in the domain. The amino acid sequence changes and the protein features are affected. Changes in the splice site also occur. Alterations are usually a single base exchange on the CDS.

Both Sift and Polyphen predict this variant to be partially damaging. They also predict it to be benign in some cases.

Experiments suggest that these variations are typical of osteoporosis susceptibility genes in the Han Chinese since there is a strong relation between osteoporotic fracture and SNPs. A sample pool of 1012 Han Chinese woman was studied to understand their relationship with bone mineral densities (BMDs) and osteoporotic fracture1. Variations in pathology may be a reason why some loci even though they are associated with BMD, are unrelated to fractures. It could also be the case that the fracture may not have been detected, that is the ability to detect those particular types of fractures could influence the results. An allele of rs7524102 in the 1p36 region is modest (1.12) and by itself not clinically useful for purposes of prediction. Since it has a frequency of over 80% in the population, however, its population attributable risk is 17%2.
References:
- Liu JM, Zhang MG, Zhao L, et al. J Clin Endocrinol Metab 95(9):E112-120, 2010.
- Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, et al. N Engl J Med 358:2355-2365, 2008.
Identification of Differential Gene Expression in Substantia Nigra and Prefrontal Area 9 Brain Tissues Associated with Parkinson’s Disease
BIOL 6150: Emily Norris, Eshaw Vidyaprakash, Krutika Satish Gaonkar, Lori Gladney
Introduction:
Parkinson’s disease is a neurodegenerative disease, resulting in movement, sleep, and cognition impairment. Although the underlying genetic basis for the disease it not known, the dopamine neurons, responsible for the release of dopamine into the central nervous system, degenerate in the substantia nigra of the brain and α-synuclein-positive Lewy bodies, an ab normal aggregate of α-synuclein presynaptic protein with unknown function, form in the neocortex, the location of the prefrontal cortex area 9. These have been found in postmortem tissues of people affected with Parkinson’s disease. Middelton, et al, studied the microarray expression profiling for both the substantia nigra and prefrontal area 9 of the brain. This was done with tissue samples from two groups of age and gender matched groups of Parkinson and control subjects. It was found that differentially expressed genes in Parkinson’s patients were involved in the mitochondrial electron transport pathway, ubiquitin-proteosomal system, and anti-apoptotic gene groups (Zhang, et al, 2005). It was also found that in patients with Parkinson’s disease, there was an underexpression of genes that control cellular bioenergetics in response to peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α). When PGC-1α is activated, there is increased expression of the subunits of the mitochondrial respiratory chain and dopaminergic neuron loss is blocked (Zheng, et al, 2010).
Datasets:
We chose to examine differentially expressed genes in patients affected with Parkinson’s disease, focusing on the substantia nigra and prefrontal area 9. Our data sets, GSE20292 and GSE20168, were taken from NCBI Gene Expression Omnibus (GEO). GSE20292 was the microarray data from the whole substantia nigra in Parkinson’s disease. GSE20168 was the microarray data from the prefrontal area 9 in Parkinson’s disease. Both datasets were from post mortem tissues of two groups of age and gender matched groups of Parkinson’s and control subjects. We chose to design our overall study based on three experimental cases. Our first experimental case was the comparison of gene expression of the substantia nigra of patients affected with Parkinson’s to the control subjects, from GSE20292. Our second experimental case was the comparison of gene expression of prefrontal area 9 of patients affected with Parkinson’s to the control subjects, from GSE20168. Our third experiment was to compare gene expression between substantia nigra and prefrontal area 9 in patients with Parkinson’s.
Methods:
We downloaded the gene expression sets from GEO and imported the data into the Biometric Research Branch (BRB) ArrayTools Add-in on Excel. In the import, we chose to use the MAS5.0 algorithm to normalize the data from the Affymetrix chips so that it could be used in further statistical tests. The MAS5.0 used the median array as a reference and excluded any genes that had a minimum fold change of 1.5 in either direction from the median. We then had to create an Experimental Descriptors sheet that contained a list of all of the sample ids. Each id was then identified, per tissue type, as either control or disease. Each id that was related to a diseased sample was also identified by the tissue it came from (for the third experiment). For each experiment, we ran a class comparison using a t-test between groups of arrays (i.e. disease versus control and substantia nigra vs prefrontal area 9) with a significance threshold of 0.001. This resulted in a list of the genes, which we then annotated, with significant expression values for each experiment.

Results of class comparison for Substantia nigra: 236 genes significant

Results of class comparison for Prefrontal Area 9: 304 genes significant

Results of class comparison for Prefrontal Area 9 vs Substantia nigra: 1787 genes significant
Volcano Plots:
Volcano plots are a graphical representations of the results of t-tests. The x-axis is the log of the fold change and the y-axis is the log of the p-value. The dotted line represents the p-value of 0.001. These plots allow for the visualization of genes with high significance – dots that are blue, above, and farther away from the p-value line are highly significant when compared with the dots that are blue, above, and closer to the p-value line. Each dot corresponds to a probeset which corresponds to a specific gene.

Substantia nigra tissue control vs disease state

Prefrontal Area 9 tissue control vs disease state

Prefrontal Area 9 tissue vs Substantia nigra
Principle Component Analysis:
In BRB ArrayTools, a multi-dimensional scaling (MDS) plot is their version of a principle component analysis when Euclidean distances are used as the distance metric. These plots show the similarity/dissimilarity between data points based on variance depending on their clustering.

Substantia nigra MDS plot – green: control, blue: disease
From the plot it is seen that the disease samples cluster together, indicating similarities in their gene expression profiles.

Prefrontal area 9 MDS plot – green: control, blue: disease
From the plot it is seen that the control samples cluster by themselves as do the disease samples. This shows clear differences in the gene expressions between the overall control samples and disease samples. The members within each sample contain similar gene expression profiles.

Prefrontal Area 9 vs Substantia nigra MDS plot – green: Prefrontal Area 9; blue: Substantia nigra
From the plot it is seen that the prefrontal area 9 samples mainly cluster by themselves as do the substantia nigra samples. This shows that the samples for the prefrontal area 9 have similar gene expression profiles as do the samples for the substantia nigra. Both groups have differing gene expressions from each other.
Hierarchical clustering:
The maps of hierarchical clustering show clustering of up-regulated and down-regulated genes for each experiment. Based on whether the samples are control, disease, or tissue-specific we are able to see that genes that are up-regulated in one state/tissue are often down-regulated in the other state/tissue. These clusters may indicate pathway up/down-regulation in Parkinson’s disease depending on whether the genes involved are from the same pathway.

Hierarchical clustering of Substantia nigra tissue control/disease state: probe sets (genes) on y-axis, samples (control/disease) on x-axis; left column: control, right column: disease
Up regulation in dark blue; Down regulation in light blue

Hierarchical clustering of prefrontal area 9 tissue control/disease state: probe sets (genes) on y-axis, samples (control/disease) on x-axis; left column: control, right column: disease
Up regulation in dark blue; Down regulation in light blue
![]()
Hierarchical clustering of substantia nigra tissue vs prefrontal area 9 tissue: probe sets (genes) on x-axis, samples (tissue) on y-axis; top row: prefrontal area 9, bottom row: substantia nigra
Pathway Analysis:
Pathway analysis was done using Database for Annotation, Visualization and Integrated Discovery (DAVID). Using the ranked probe set lists created from our class comparison experiments, we used the functional annotation tool to determine what pathways contained our differentially expressed genes.
A functional annotation chart comparing the genes with their related gene ontology (GO) terms shows that many of the differentially expressed genes are related to cell signaling, the cell cycle, and regulation of cell death (apoptosis). These genes were found to be significant in all of our experiments, indicating that they could be associated with Parkinson’s disease.

Functional Annotation Chart
In agreement with the findings of the original study, the majority of the differentially expressed genes in all three experiments were found to be involved in pathways relating to oxidative phosphorylation (electron transport in the mitochondria), proteasome ubiquitination, and apoptosis.

KEGG Pathway of mitochondrial oxidative phosphorylation

KEGG Pathway of the proteasome and ubiquitination.

KEGG Pathway of the cell cycle and apoptosis.
We also found that the significant genes were involved in the pathways relating to Alzheimer’s disease, neuronal signaling, and cell signaling.

KEGG Pathway of Alzheimer’s disease.

KEGG Pathway of neurotrophin signaling.

KEGG Pathway of long term potentiation.

KEGG Pathway of long term depression.
The above significant pathways were found in all three of our experiments. Although there were other pathways related to the significant probesets/genes per experiment, there were very few genes involved in the pathways, indicating that these are not important to our study of Parkinson’s. We felt it important to explore the pathways that involved the most number of significant genes, deeming those pathways as the ones associated with the disease.
Discussion:
Although the cause of Parkinson’s disease is unknown, increased oxidative stress and mitochondrial dysfunction in the dopamine neurons are central to the disease. There is also a deficit in mitochondrial complex 1 which may arise from or contribute to increased cellular oxidative stress.
The mitochondrial genes were differentially expressed in the disease states and the normal state comparison in the substantia nigra and prefrontal tissues in the brain. If there is a dysfunction in these genes might lead to ATP depletion in the neurons which lead to the degeneration receptors and inefficient signalling through the neurons.
We found that the gene lists from the control /disease of the two tissues were enriched in the Alzheimer’s disease pathway and apoptosis as expected. The long term depression and long term potentiation pathways are important part of the manipulation of the motor function and coordination. Thus a difference in expression of the genes involved in these pathways may be a possible cause for movement disorders.
As seen from the pathway analysis, cell death is caused when there is an ATP depletion in the cell through complex (1,2,3,4,5)deficiency in the mitochondria. These organelles coordinate energy metabolism and are responsible for the generation of the majority of cellular ATP. Neurons, as metabolically active cells with high energy demands, are predominantly dependent on mitochondrial function, as reflected by the observation that mitochondrial defects are key features of chronic neurodegenerative diseases.
Most drug treatments increase the level of dopamine in the brain or oppose the action of acetylcholine. Levodopa which is a precursor for dopamine is widely used to compensate for the dopamine loss in the Parkinson’s patients. The drug is useful for the initial stages of the disease but as the disease progresses the drug becomes less effective. The patient may also experience some side effects such as increase in involuntary actions and dyskinesia which is one of the main problems. Therefore there is a need for dopamine independent drugs. The reduced behavioural deficits following dopamine depletion in RGS4 deficient mice indicate that RGS4 inhibition may be an effective non-dopamine dependent strategy for treating Parkinson’s disease.

Volcano plot: RGS4 statistically significant gene
As seen in the above volcano plot, the probe set for RGS4 can be considered to have highly significant expression at a p-value of 0.001. This allows us to conclude that this gene may be a drug target in patients with Parkinson’s disease.
The Regulator of G protein (RGS) is a group of protein that regulates the life time of the active G alpha-GTP by accelerating the GTP hydrolysis. RGS4 is expressed strongly in MSNs in the dorsolateral striatum, where it regulates the activity of mGluR5 and PLCb (Gold et al., 1997; Schwendt and McGinty, 2007), its activity is increased by PKA phosphorylation (Huang et al., 2007), and it strongly inhibits signaling through Gq (Saugstad et al., 1998). Loss of RGS4 in direct-pathway MSNs and cholinergic interneurons (Ding et al., 2006) may also be contributing to the effects of dopamine depletion since RGS4 is expressed in all of these cell types (Taymans et al., 2004).
References:
1. Zhang Y, James M, Middleton FA, Davis RL. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet 2005 Aug 5;137B(1):5-16. PMID: 15965975
2. Zheng B, Liao Z, Locascio JJ, Lesniak KA et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2010 Oct 6;2(52):52ra73. PMID: 20926834
3. Lerner, T.N., Horne, E.A., Stella, N., and Kreitzer, A.C. (2010) Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J. Neurosci. 30,2160–2164
4. Ahn, K., McKinney, M.K., and Cravatt, B.F. (2008) Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 108, 1687–1607.
5. Piomelli, D. (2003)The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4, 873–884.
6. Gold, S.J., Ni, Y.G., Dohlman, H.G., and Nestler, E.J. (1997) Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J. Neurosci. 17, 8024–8037.
7. Schwendt, M., and McGinty, J.F. (2007) Regulator of G-protein signaling 4 interacts with metabotropic glutamate receptor subtype 5 in rat striatum: relevance to amphetamine behavioral sensitization. J. Pharmacol. Exp. Ther. 323, 650–657.
8. Ding, J., Guzman, J.N., Tkatch, T., Chen, S., Goldberg, J.A., Ebert, P.J., Levitt, P., Wilson, C.J., Hamm, H.E., and Surmeier, D.J. (2006) RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat. Neurosci. 9, 832–842.
9. Taymans, J.M., Kia, H.K., Claes, R., Cruz, C., Leysen, J., and Langlois, X. (2004) Dopamine receptor-mediated regulation of RGS2 and RGS4 mRNA differentially depends on ascending dopamine projections and time. Eur. J. Neurosci. 19, 2249–2260.
10. Huang, J., Zhou, H., Mahavadi, S., Sriwai, W., and Murthy, K.S. (2007) Inhibition of Galphaq-dependent PLC-beta1 activity by PKG and PKA is mediated by phosphorylation of RGS4 and GRK2. Am. J. Physiol. Cell Physiol. 292, C200–C208.
11. Saugstad, J.A., Marino, M.J., Folk, J.A., Hepler, J.R., and Conn, P.J. (1998) RGS4 inhibits signaling by group I metabotropic glutamate receptors. J. Neurosci. 18, 905–913.
12. Lerner T. N. and Kreitzer A. C. (2012) RGS4 Is Required for Dopaminergic Control of Striatal LTD and Susceptibility to Parkinsonian Motor Deficits. Neuron. 73, 347–359
